Apply the estimates from cycle_npreg_insample
to another gene expression dataset to infer an angle, or cell cycle
phase, for each cell.
cycle_npreg_outsample( Y_test, sigma_est, funs_est, method.trend = c("trendfilter", "loess", "bspline"), normed = TRUE, polyorder = 2, method.grid = "uniform", ncores = 2, grids = 100, get_trend_estimates = FALSE )
Arguments
| Y_test | A |
|---|---|
| sigma_est | A vector of gene-specific standard errors of the cyclic trends. |
| funs_est | A vector of cyclic functions used to estimate cyclic trends. |
| method.trend | How to estimate cyclic trend of gene expression
values. We offer three options: |
| normed | Is the data already normalized? |
| polyorder | We estimate cyclic trends of gene expression levels using nonparamtric trend filtering. |
| method.grid | The approach used to initialize angles in the
computation. |
| ncores | We use doParallel package for parallel computing. |
| grids | number of bins to be selected along 0 to 2pi. |
| get_trend_estimates | Re-estimate the cylic trend based on the
predicted cell cycle phase, or not ( |
Value
A list with the following elements:
The inputted gene expression marix.
Inferred angles or cell cycle phases (not ordered).
Log-likelihoods for each gene.
The inferred angles, in ascending order.
The input gene expression matrix reordered by
cell_times_reordered.
Standard error of the cyclic trend for each
gene, reordered by cell_times_reordered.
A list of functions for approximating the
cyclic trends of gene expression levels for each gene, reordered by
cell_times_reordered.
Estimated cyclic trend of gene expression
values for each gene, reordered by cell_times_reordered.
Probabilities of each cell belonging to each bin.
See also
cycle_npreg_insample for estimating parameters for
cyclic functions from training data,
cycle_npreg_loglik for log-likehood at angles between
0 to 2pi, and cycle_npreg_mstep for estimating cyclic
functions given inferred phases from
cycle_npreg_loglik.
Examples
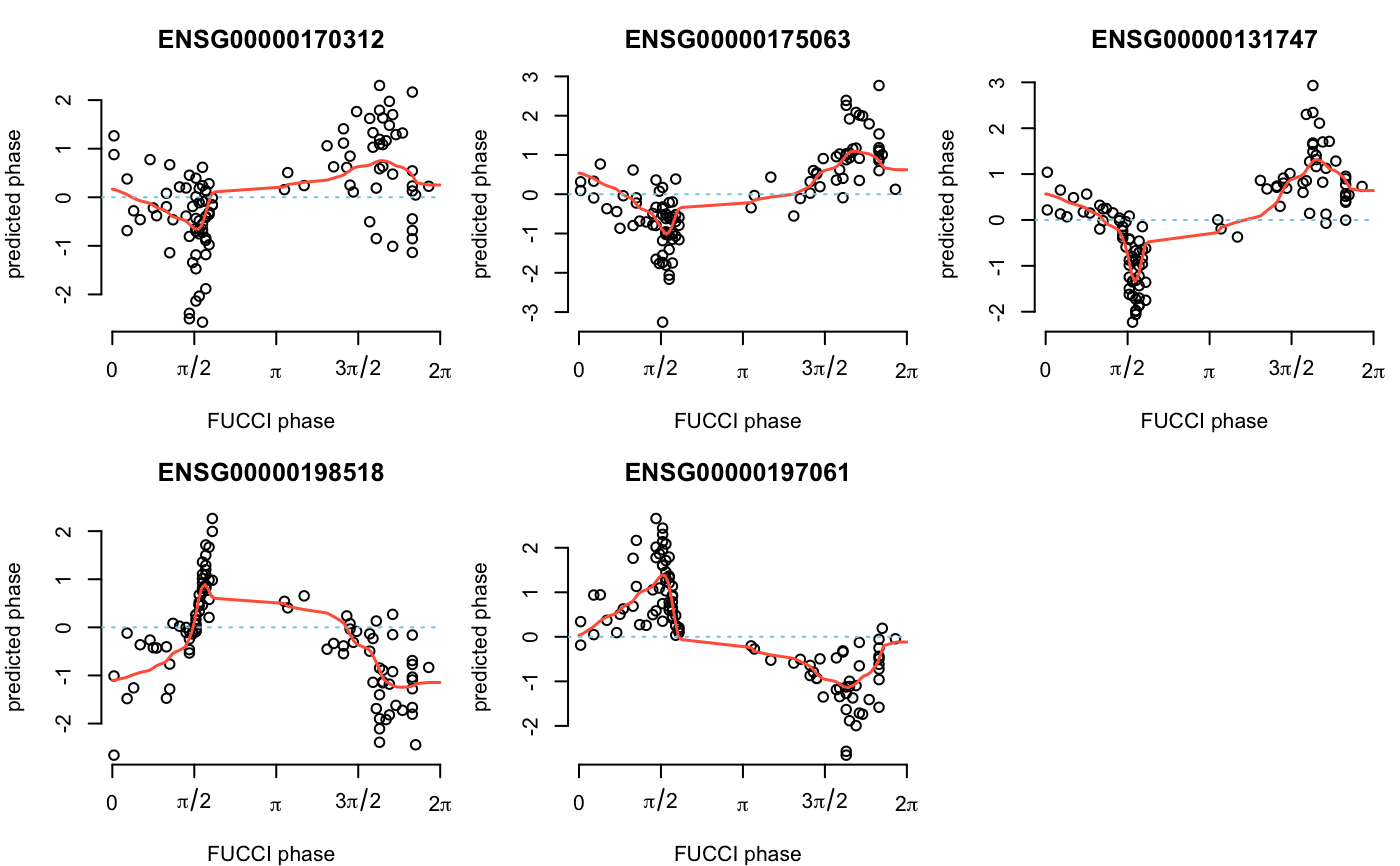
library(SingleCellExperiment)#>#>#>#>#>#> #>#> #> #> #> #>#> #> #>#> #> #> #> #> #> #> #>#>#> Warning: package ‘S4Vectors’ was built under R version 3.6.3#> #>#> #> #>#>#>#> Warning: package ‘GenomeInfoDb’ was built under R version 3.6.3#>#> #> #> #> #>#>#> Warning: package ‘DelayedArray’ was built under R version 3.6.3#>#> #>#> #> #>#>#> #>#> #> #>#> #> #>data(sce_top101genes) # Select top 5 cyclic genes. sce_top5 <- sce_top101genes[order(rowData(sce_top101genes)$pve_fucci, decreasing = TRUE)[1:5],] # Select NA18511 samples for training and test sets. coldata <- colData(sce_top5) which_samples_train <- rownames(coldata)[coldata$chip_id != "NA18511"] which_samples_predict <- rownames(coldata)[coldata$chip_id == "NA18511"] # Learn gene cyclic functions from the training data using the # trend filtering method. sce_top5 <- data_transform_quantile(sce_top5)#>expr_quant <- assay(sce_top5, "cpm_quantNormed") Y_train <- expr_quant[, colnames(expr_quant) %in% which_samples_train] theta_train <- coldata$theta_shifted[rownames(coldata) %in% which_samples_train] names(theta_train) <- rownames(coldata)[rownames(coldata) %in% which_samples_train] model_5genes_train <- cycle_npreg_insample(Y = Y_train,theta = theta_train, polyorder = 2,ncores = 2, method.trend = "trendfilter")#># Predict cell cycle in the test samples. Y_test <- sce_top5[,colnames(sce_top5) %in% which_samples_predict] model_5genes_predict <- cycle_npreg_outsample(Y_test = Y_test, sigma_est = model_5genes_train$sigma_est, funs_est = model_5genes_train$funs_est, method.trend = "trendfilter", ncores = 2,get_trend_estimates = FALSE) # Estimate cyclic gene expression levels given the cell cycle for each # gene. predict_cyclic <- fit_cyclical_many(Y = assay(model_5genes_predict$Y,"cpm_quantNormed"), theta = colData(model_5genes_predict$Y)$cellcycle_peco)#># Plot the cell cycle predictions vs. FUCCI phase. par(mfrow = c(2,3),mar = c(4,4,3,1)) for (g in seq_along(rownames(model_5genes_predict$Y))) { plot(assay(model_5genes_predict$Y,"cpm_quantNormed")[ rownames(model_5genes_predict$Y)[g],], x = colData(model_5genes_predict$Y)$cellcycle_peco,axes = FALSE, xlab = "FUCCI phase",ylab = "predicted phase") x <- seq(0,2*pi,length.out = 100) lines(x = x,y = predict_cyclic$cellcycle_function[[ rownames(model_5genes_predict$Y)[g]]](x), lwd = 1.5,col = "tomato") axis(2) axis(1,at = seq(0,2*pi,length.out = 5), labels = c(0,expression(pi/2),expression(pi),expression(3*pi/2), expression(2*pi))) abline(h = 0,lwd = 1,col = "skyblue",lty = "dotted") title(names(model_5genes_predict$Y)[g]) }