Choosing number of clusters in GoM model
Joyce Hsiao
Warning in as.POSIXlt.POSIXct(Sys.time()): unknown timezone 'zone/tz/2018c.
1.0/zoneinfo/America/Chicago'Last updated: 2018-06-07
Code version: 852b84b
Background and summar
The GoM (Grades of Membership) model computes log Bayes factor for comparing model fitting with k clusters versus null single cluster model. The log Bayes factor is corrected for the number of parameters fitted in each model. In clustering analysis, one typically chooses k that has the largest Bayes factor. However, it is worth remembering that in practice, it is unlikely to get a true k, and results from different k may complement each other rather than competing with each other.
In the current analysis, we evaluated the data with k = 2 to 20. We found that k=2 produces the largest Bayes factor.
Analysis
library(knitr)
library(kableExtra)
library(dplyr)
library(metagenomeSeq)
library(CountClust)
library(vegan)Read in filtered data.
MRobj <- readRDS("../data/nasal_filtered_normed_batchcorrected.rds")
counts <- MRcounts(MRobj,norm=FALSE,log=FALSE)
MRobjMRexperiment (storageMode: environment)
assayData: 553 features, 197 samples
element names: counts
protocolData: none
phenoData
sampleNames: EM0042 EM0047 ... E0168 (197 total)
varLabels: StudyID age ... infnone (94 total)
varMetadata: labelDescription
featureData
featureNames:
k__Bacteria;p__[Thermi];c__Deinococci;o__Deinococcales;f__Deinococcaceae;g__CM44;s__
k__Bacteria;p__[Thermi];c__Deinococci;o__Deinococcales;f__Deinococcaceae;g__Deinococcus;s__
... s__zeae (553 total)
fvarLabels: Kingdom Phylum ... Species (7 total)
fvarMetadata: labelDescription
experimentData: use 'experimentData(object)'
Annotation: Fitting k=2 to 10.
fits <- FitGoM(t(counts), K=c(2:10), tol=0.00001)
saveRDS(fits, file = "../data/gomfits.rds")Extract sample memberships.
fits <- readRDS(file = "../data/gomfits.rds")
clust_fit_membership <- lapply(fits, function(xx) {
apply(xx$omega, 1, which.max)
})
for (i in 1:length(clust_fit_membership)) {
print(table(clust_fit_membership[[i]]))
}
1 2
180 17
1 2 3
166 24 7
1 2 3 4
88 71 27 11
1 2 3 4 5
105 68 15 8 1
1 2 3 4 5 6
73 54 40 17 12 1
1 2 3 4 5 6 7
75 42 38 29 7 3 3
1 2 3 4 5 6 7 8
79 48 26 19 12 9 2 2
1 2 3 4 5 6 7 8 9
76 50 25 19 12 9 2 3 1
1 2 3 4 5 6 7 8 9 10
79 30 27 16 18 11 7 2 5 2 Log Bayes Facctor
par(mfrow=c(1,2))
plot(bf_species, type = "b", xlab = "Number of clusters",
ylab = "log Bayes Factor (H1 vs single cluster)", main = "Species")
plot(bf_species_seq, type = "b", xlab = "Number of clusters",
ylab = "Percent change in log Bayes Factor (k vs k-1)", main = "Species")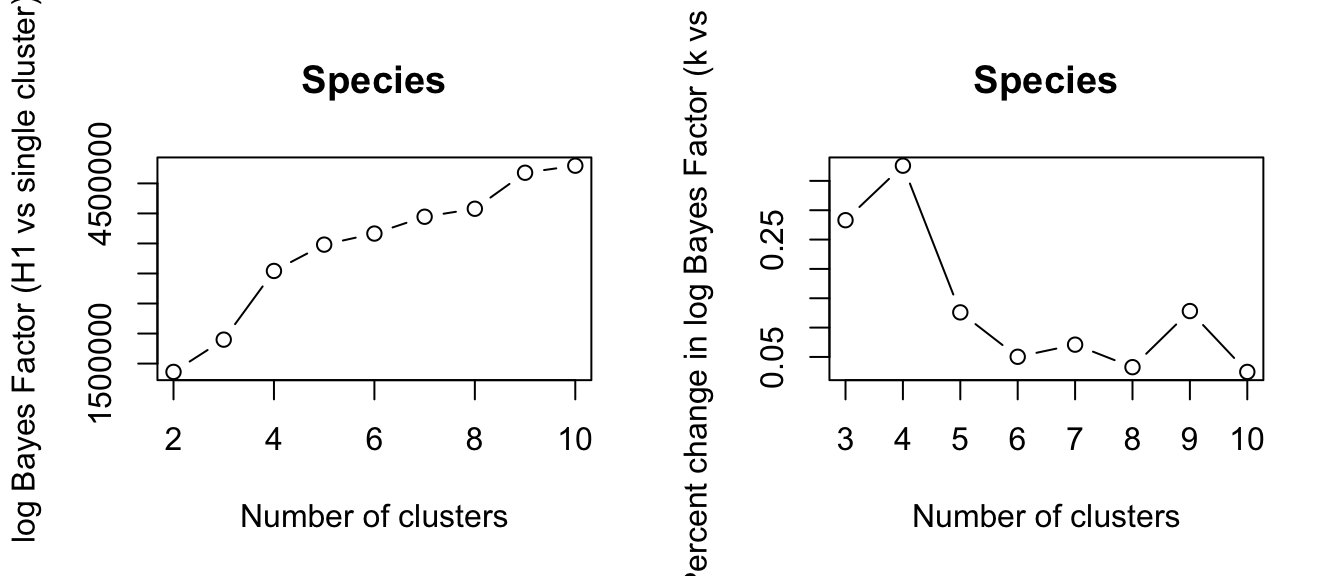
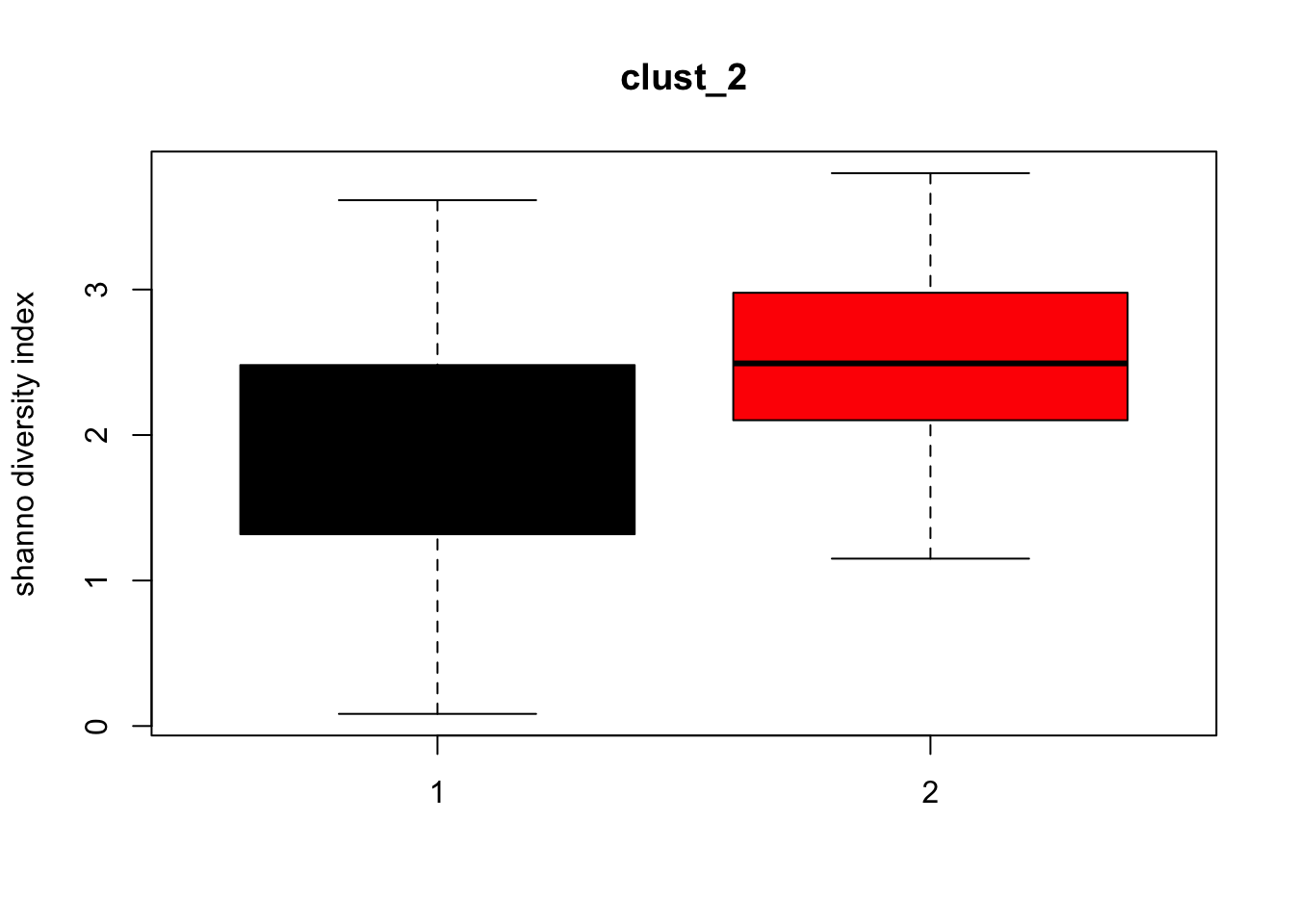
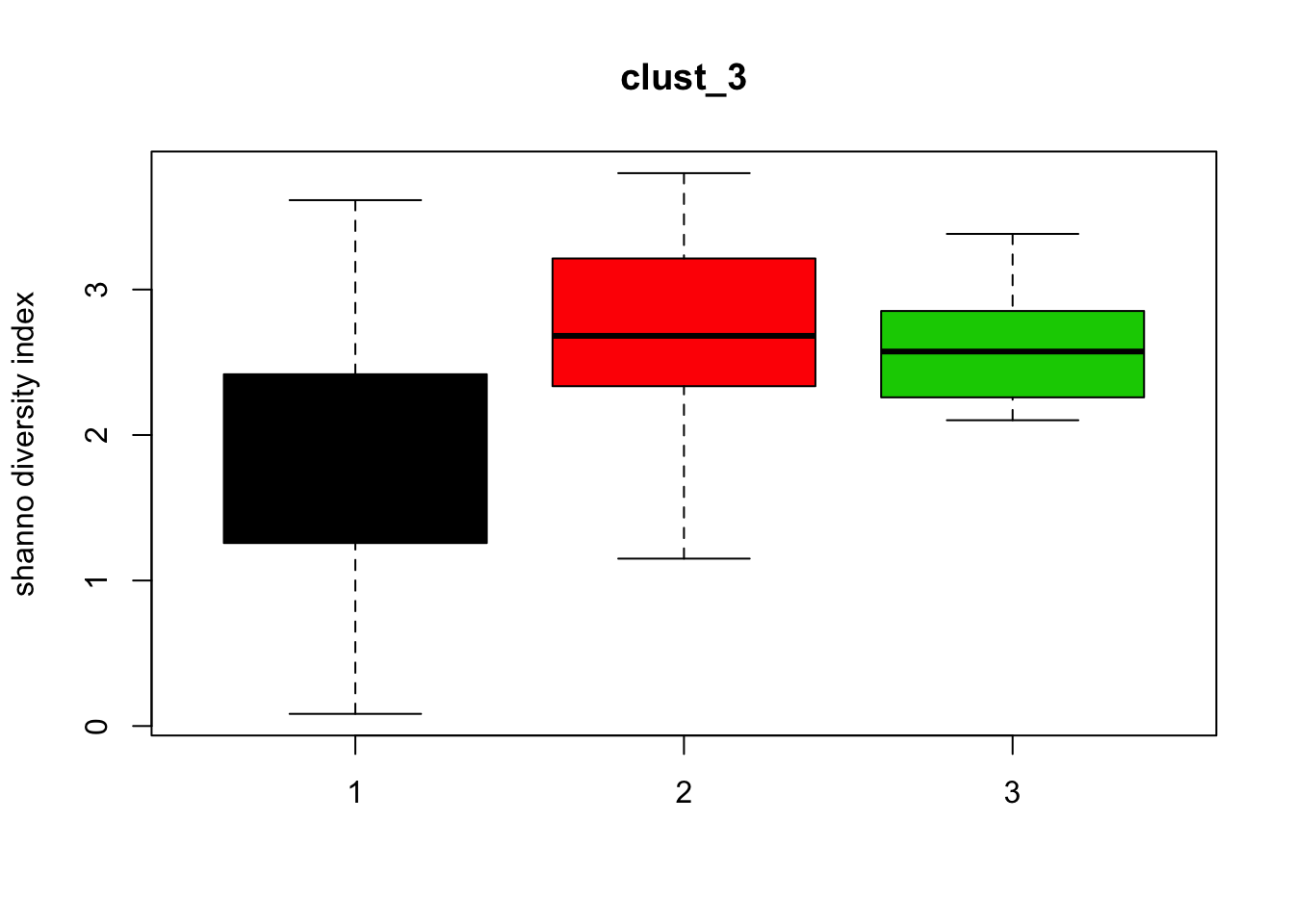
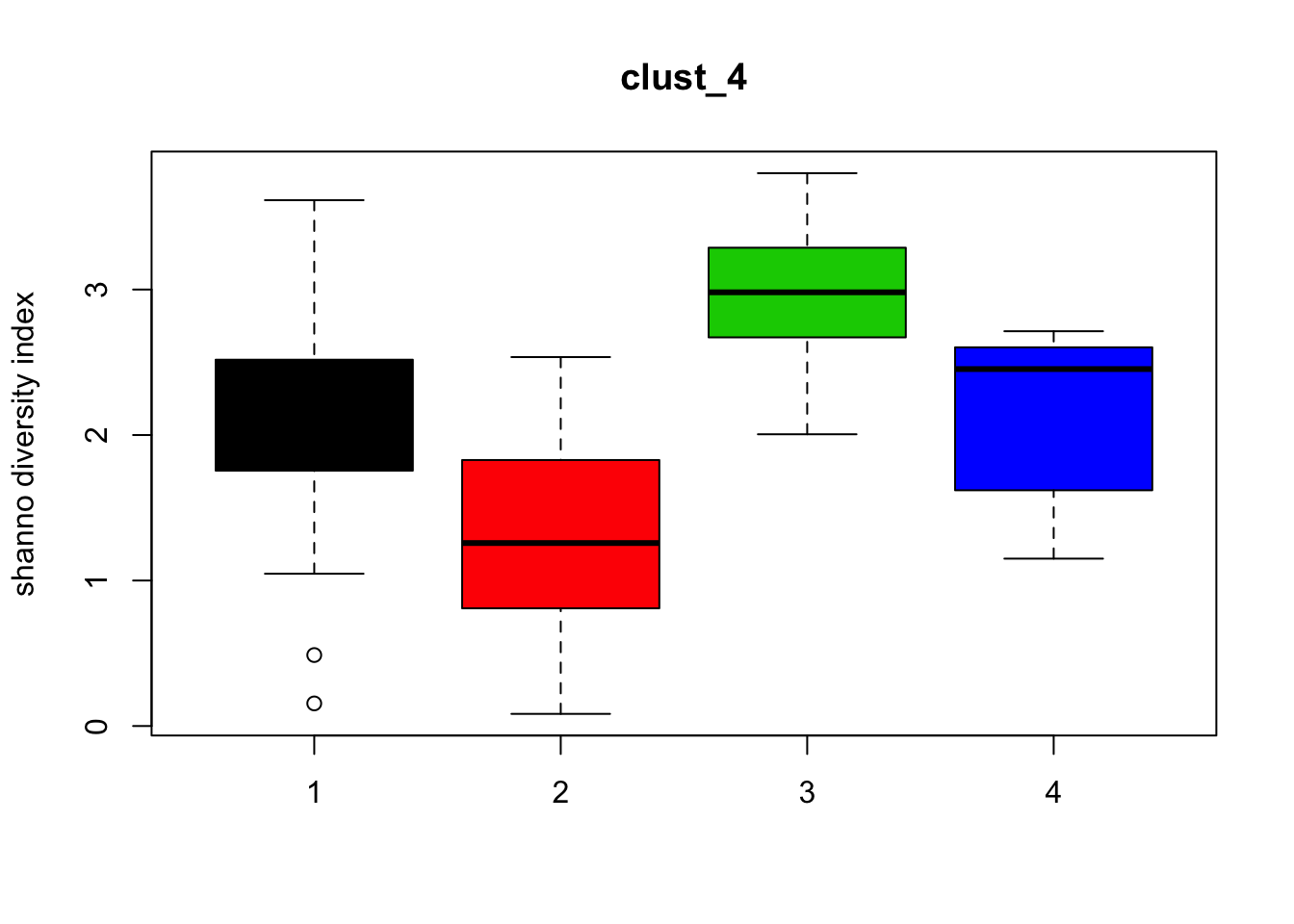
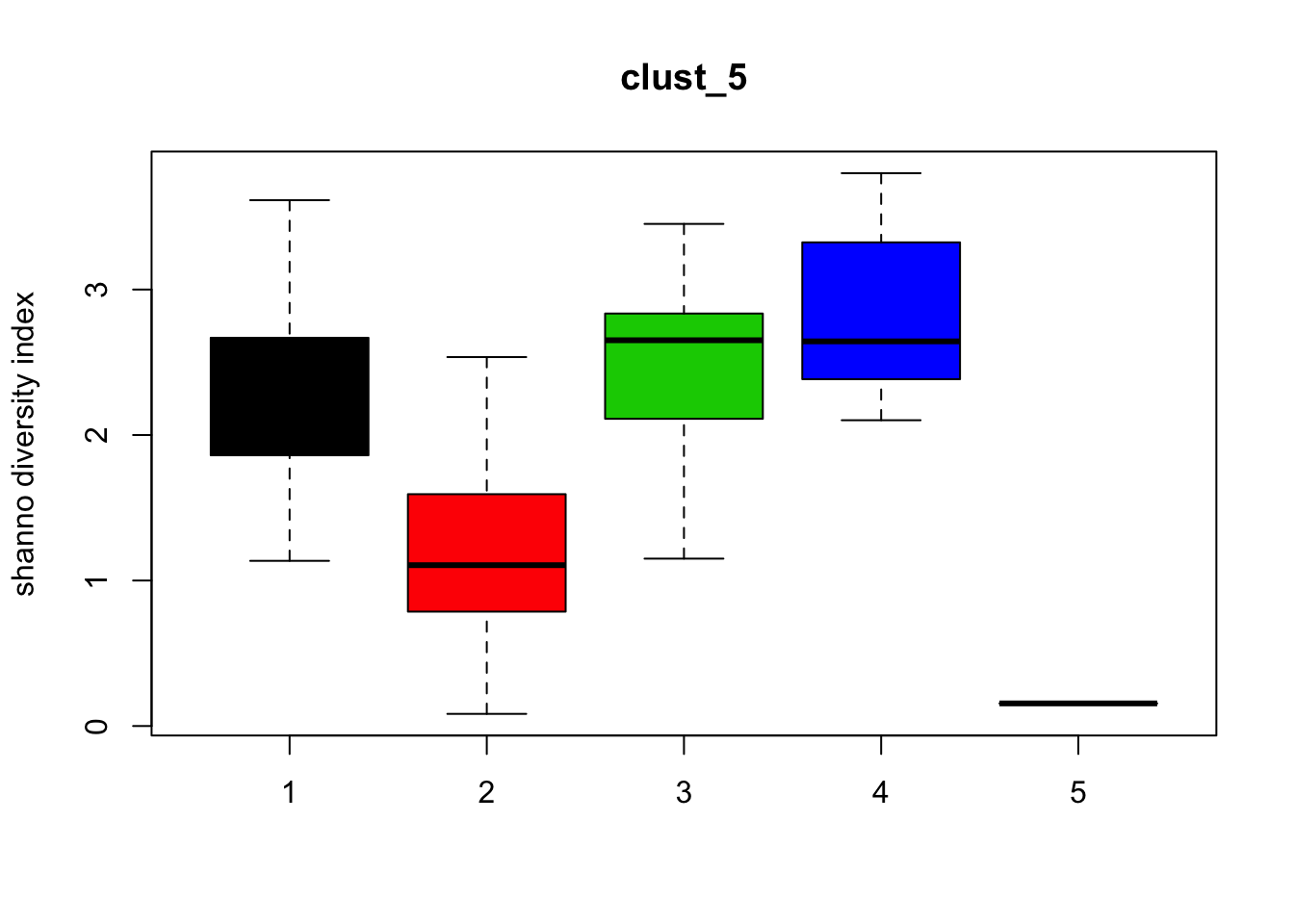
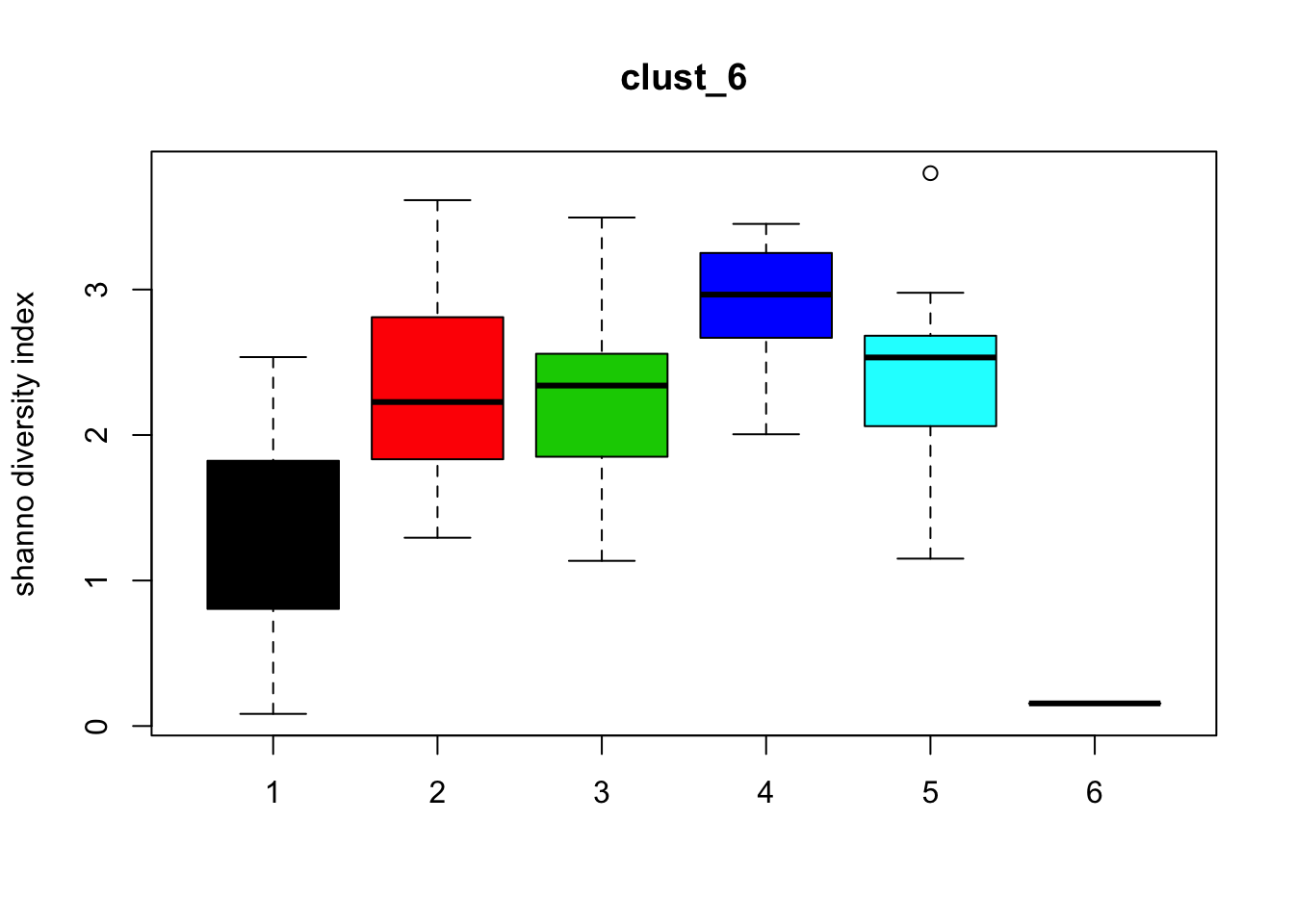
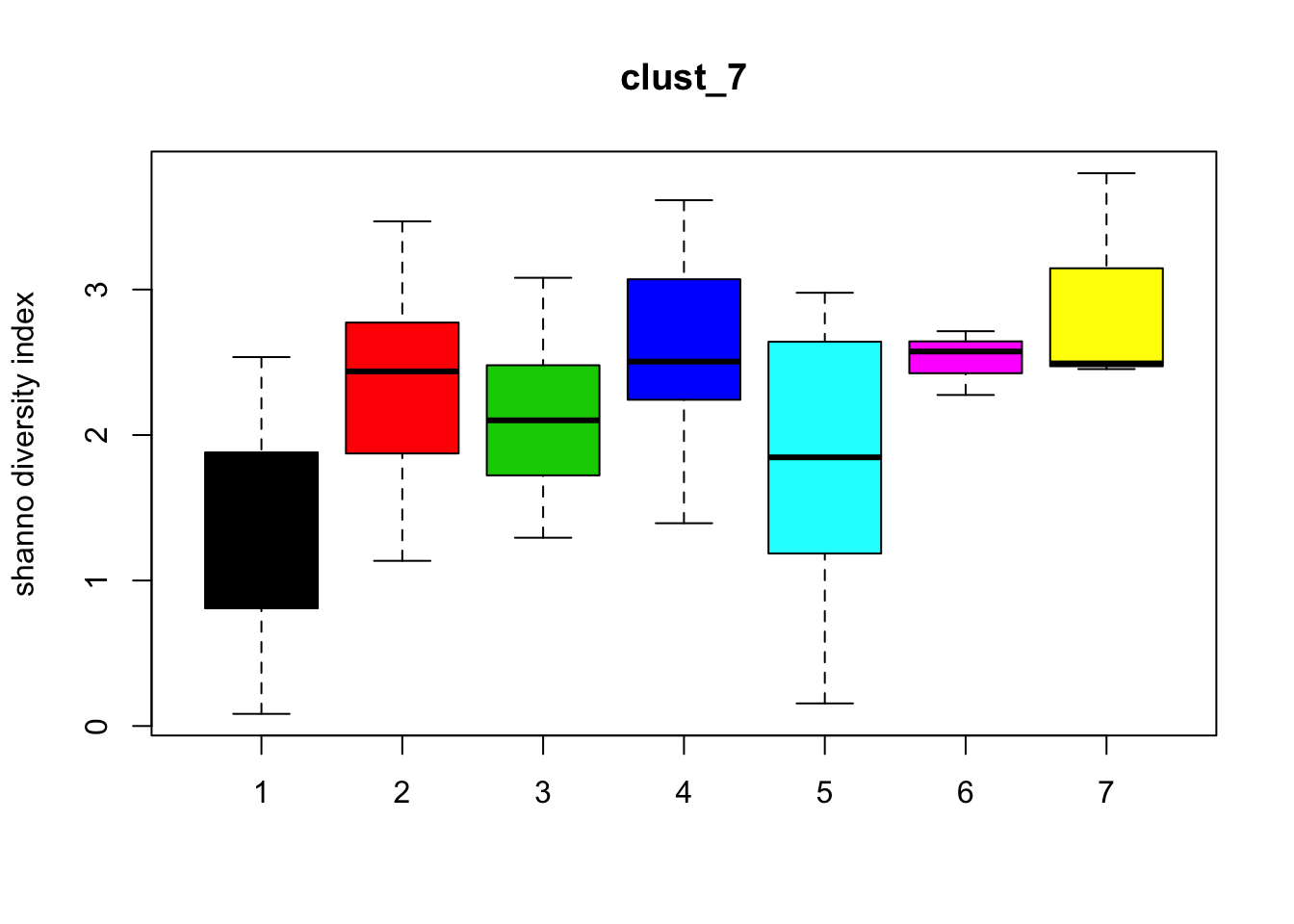
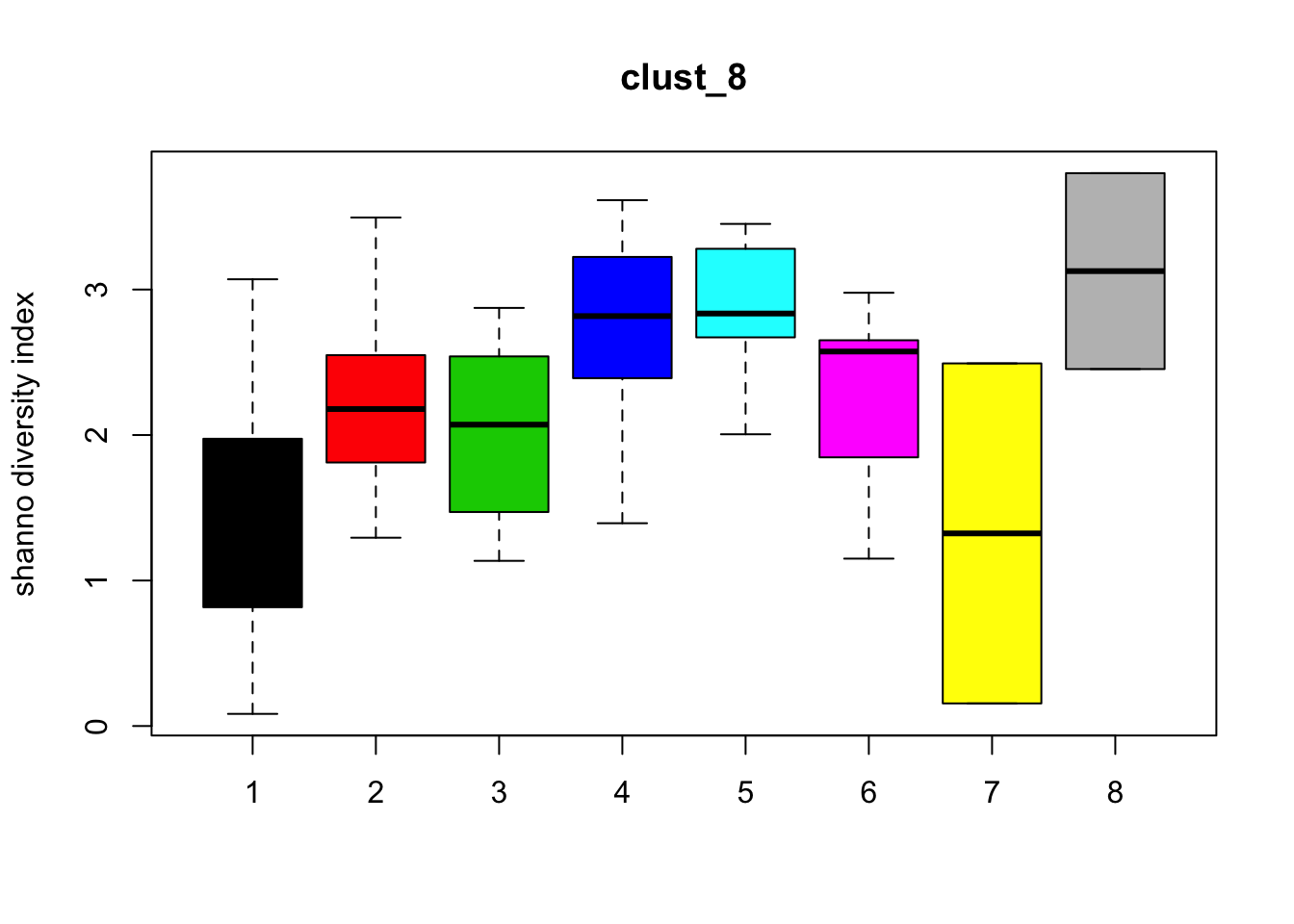
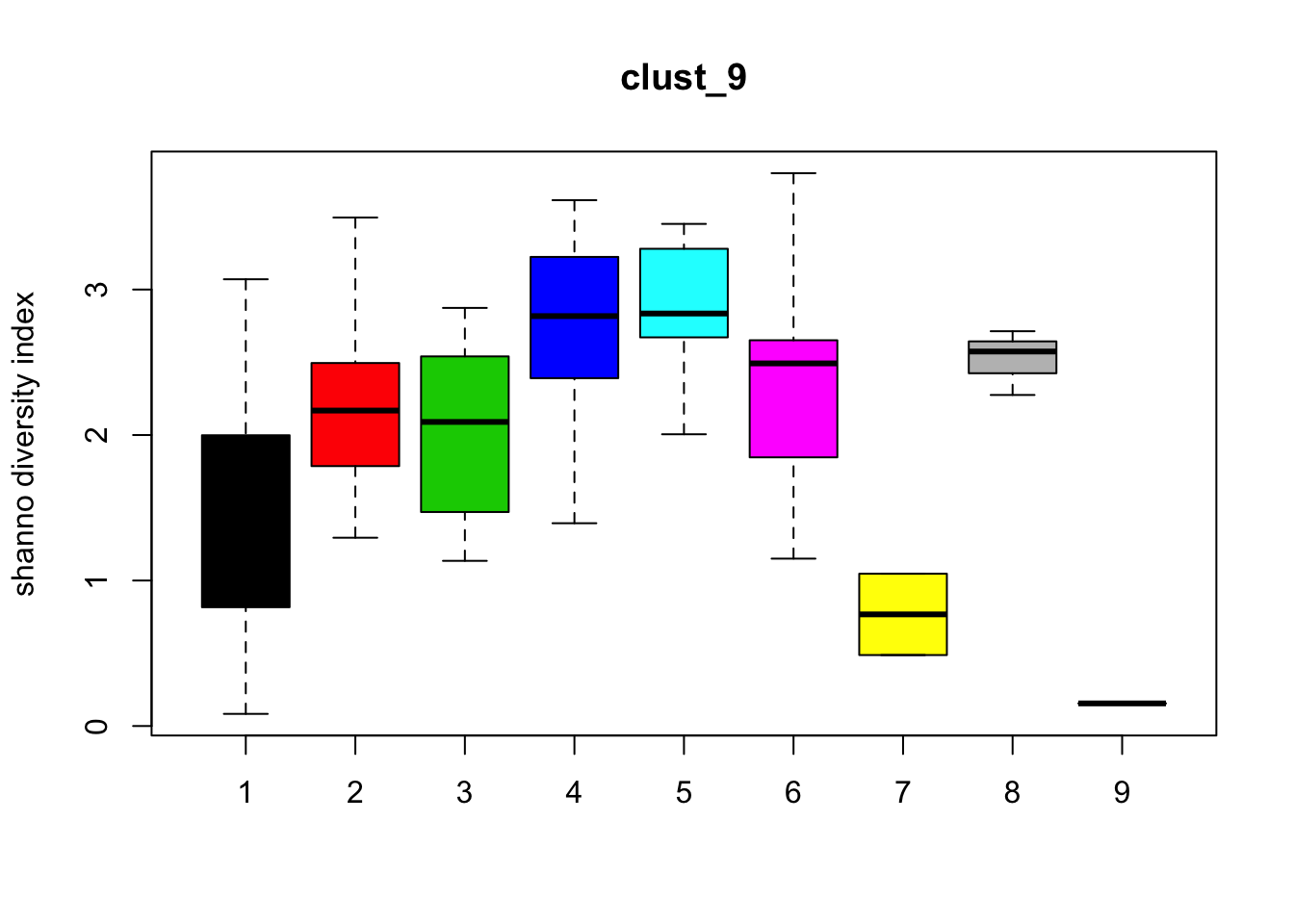
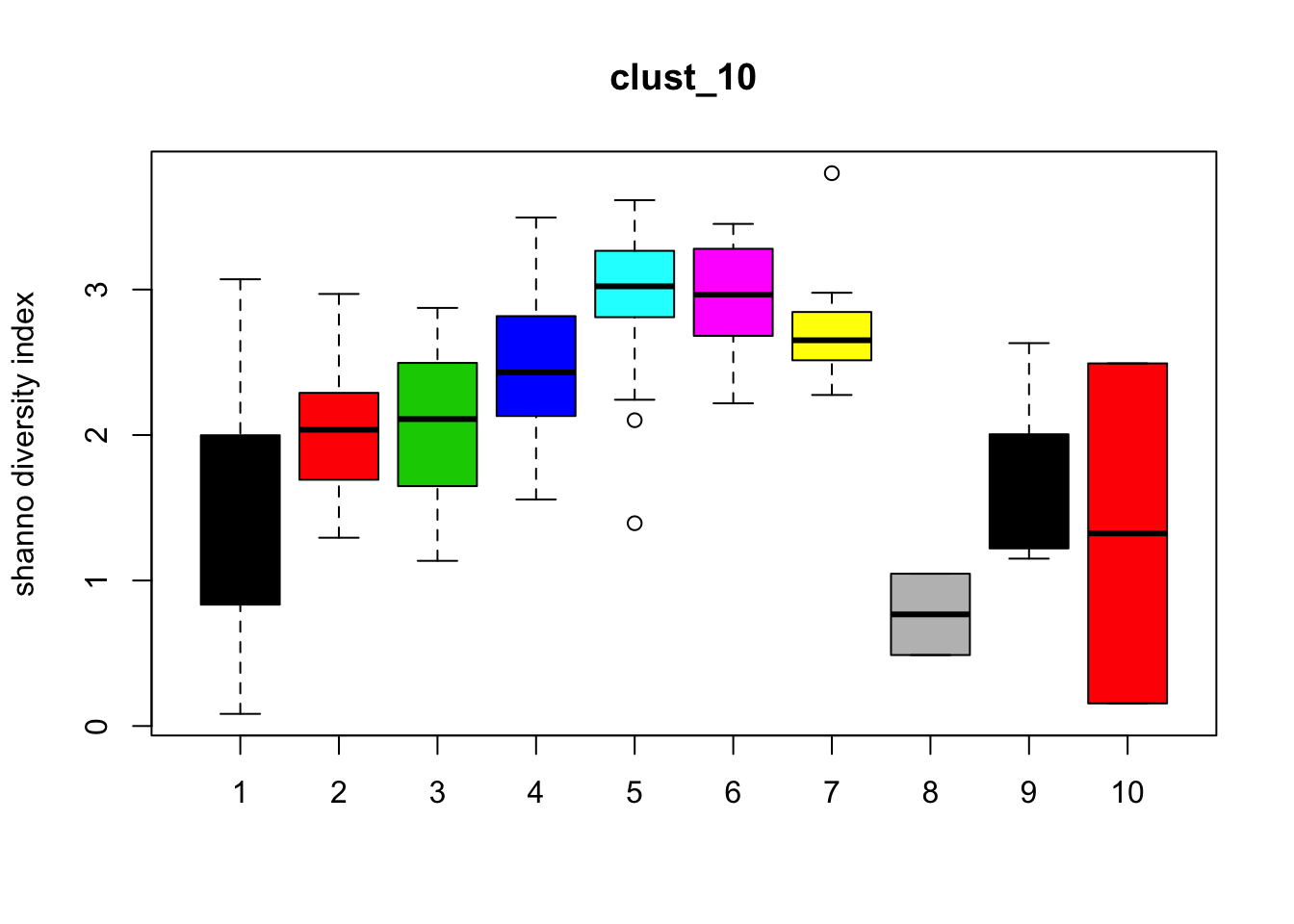
Diversity
for (i in 1:length(clust_fit_membership)) {
cl <- factor(clust_fit_membership[[i]])
mat <- t(MRcounts(MRobj,norm=FALSE,log=FALSE))
H <- diversity(mat,index="shannon")
boxplot(H~cl, ylab=paste("shanno","diversity index"), col=1:length(levels(cl)),
main = names(clust_fit_membership)[i])
}
Session information
R version 3.4.1 (2017-06-30)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS High Sierra 10.13
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.4/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.4/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] parallel stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] vegan_2.5-1 lattice_0.20-35 permute_0.9-4
[4] CountClust_1.4.1 ggplot2_2.2.1 metagenomeSeq_1.21.1
[7] RColorBrewer_1.1-2 glmnet_2.0-16 foreach_1.4.4
[10] Matrix_1.2-14 limma_3.34.9 Biobase_2.38.0
[13] BiocGenerics_0.24.0 dplyr_0.7.4 kableExtra_0.8.0
[16] knitr_1.20
loaded via a namespace (and not attached):
[1] httr_1.3.1 viridisLite_0.3.0 gtools_3.5.0
[4] assertthat_0.2.0 stats4_3.4.1 yaml_2.1.19
[7] slam_0.1-43 pillar_1.2.2 backports_1.1.2
[10] glue_1.2.0 digest_0.6.15 rvest_0.3.2
[13] colorspace_1.3-2 picante_1.6-2 cowplot_0.9.2
[16] htmltools_0.3.6 plyr_1.8.4 pkgconfig_2.0.1
[19] scales_0.5.0 gdata_2.18.0 git2r_0.21.0
[22] tibble_1.4.2 mgcv_1.8-23 nnet_7.3-12
[25] lazyeval_0.2.1 magrittr_1.5 evaluate_0.10.1
[28] nlme_3.1-137 MASS_7.3-50 gplots_3.0.1
[31] xml2_1.2.0 tools_3.4.1 hms_0.4.2
[34] matrixStats_0.53.1 stringr_1.3.0 munsell_0.4.3
[37] cluster_2.0.7-1 bindrcpp_0.2.2 compiler_3.4.1
[40] caTools_1.17.1 rlang_0.2.0 grid_3.4.1
[43] iterators_1.0.9 rstudioapi_0.7 bitops_1.0-6
[46] rmarkdown_1.9 boot_1.3-20 gtable_0.2.0
[49] codetools_0.2-15 flexmix_2.3-14 reshape2_1.4.3
[52] R6_2.2.2 bindr_0.1.1 rprojroot_1.3-2
[55] maptpx_1.9-3 KernSmooth_2.23-15 readr_1.1.1
[58] ape_5.1 modeltools_0.2-21 stringi_1.2.2
[61] SQUAREM_2017.10-1 Rcpp_0.12.16 This R Markdown site was created with workflowr